Leveraging Novel Information Sources for Protein Structure Prediction
Michael Bohlke-Schneider
Title: Leveraging Novel Information Sources for Protein Structure Prediction
Abstract:
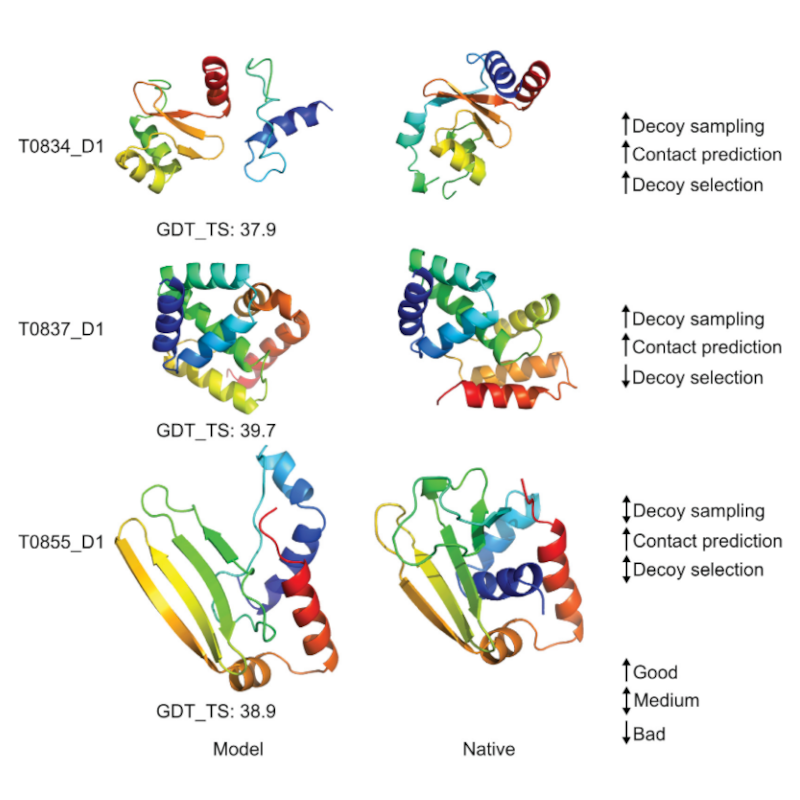
Three-dimensional protein structures are an invaluable stepping stone towards the understanding of cellular processes. Computational protein structure prediction holds the promise of providing these structural models at low cost and effort. However, the major bottleneck towards effective protein structure prediction is the high dimensionality and vast size of the protein conformational space. These properties of the conformational space make it extremely difficult to locate the native structure through search. Information alleviates this issue by guiding search towards the native protein structure. Thus, information is invaluable in conformational space search. Not surprisingly, state-of-the-art structure prediction methods heavily rely on information. Obviously, unlocking novel sources of information should further increase our ability to accurately predict protein structure. This thesis leverages three novel sources of information to advance protein structure prediction. First, we leverage physicochemical information that is encoded in energy functions and predicted structure models. Native contact networks form characteristic patterns to be energetically favorable. This thesis develops a network-based representation to capture these patterns and uses this representation to predict residue-residue contacts. The second source of information is experimental data from high-density cross-linking/ mass spectrometry (CLMS) experiments. We integrate this information in an experimental/computational hybrid method for protein structure determination. The third information source is corroborating information. Corroborating information judges the likelihood of the co-occurence of structural constraints. Nearly all methods provide these constraints in isolation, thereby neglecting any corroborating evidence between them. We develop a network-based analysis method to refine structure constraints with corroborating information. We demonstrate the value of these information sources in extensive ab initio structure prediction experiments with a customized conformational space search algorithm and a novel structure prediction pipeline. This pipeline reached state-of-the-art contact and ab initio structure prediction performance in the 11th community-wide Critical Assessment of Protein Structure Prediction experiment (CASP11).
Using our CLMS-based hybrid method, we reconstruct the domain structures of human serum albumin in solution and in its native environment, human blood serum. This represents a disruptive first step towards a mass spectrometry-driven, ab initio structure determination method that is able to probe protein structure where it really matters: In their natural environment, which is their very place of action.
December 2015