Leveraging Novel Information Sources for Protein Structure Prediction
Michael Bohlke-Schneider
Titel: Leveraging Novel Information Sources for Protein Structure Prediction
Zusammenfassung:
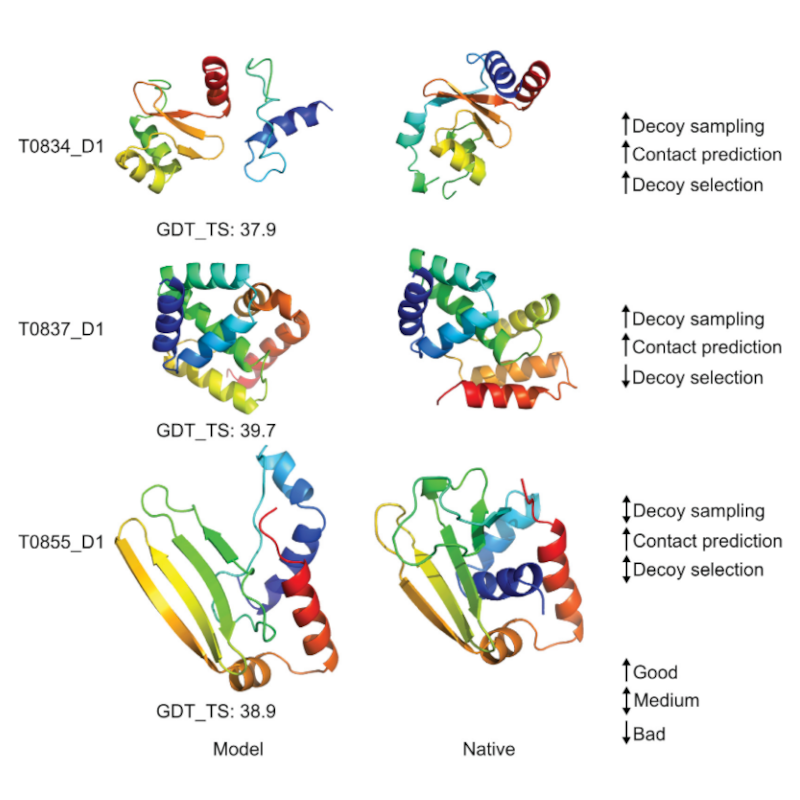
Dreidimensionale Proteinstrukturen sind ein unschätzbarer Schritt zum Verständnis zellulärer Prozesse. Die computergestützte Vorhersage von Proteinstrukturen verspricht, diese Strukturmodelle mit geringem Kosten- und Arbeitsaufwand bereitzustellen. Der größte Engpass bei der effektiven Vorhersage von Proteinstrukturen ist jedoch die hohe Dimensionalität und die enorme Größe des Konformationsraums von Proteinen. Diese Eigenschaften des Konformationsraums machen es extrem schwierig, die native Struktur durch Suche zu finden. Informationen lindern dieses Problem, indem sie die Suche in Richtung der nativen Proteinstruktur lenken. Daher sind Informationen bei der Suche im Konformationsraum von unschätzbarem Wert. Es überrascht nicht, dass die modernsten Methoden zur Strukturvorhersage stark auf Informationen angewiesen sind. Es liegt auf der Hand, dass die Erschließung neuer Informationsquellen unsere Fähigkeit zur genauen Vorhersage der Proteinstruktur weiter verbessern sollte. In dieser Arbeit werden drei neue Informationsquellen genutzt, um die Vorhersage von Proteinstrukturen zu verbessern. Erstens nutzen wir physiko-chemische Informationen, die in Energiefunktionen und vorhergesagten Strukturmodellen kodiert sind. Native Kontaktnetzwerke bilden charakteristische Muster, die energetisch vorteilhaft sind. In dieser Arbeit wird eine netzwerkbasierte Darstellung entwickelt, um diese Muster zu erfassen und diese Darstellung zur Vorhersage von Rest-Rest-Kontakten zu nutzen. Die zweite Informationsquelle sind experimentelle Daten aus hochdichten Quervernetzungs-/Massenspektrometrie-Experimenten (CLMS). Wir integrieren diese Informationen in eine experimentelle/computergestützte Hybridmethode zur Bestimmung der Proteinstruktur. Die dritte Informationsquelle sind bestätigende Informationen. Bestätigende Informationen beurteilen die Wahrscheinlichkeit des gleichzeitigen Auftretens von strukturellen Einschränkungen. Nahezu alle Methoden stellen diese Einschränkungen isoliert dar und vernachlässigen dabei jegliche bestätigende Beweise zwischen ihnen. Wir entwickeln eine netzwerkbasierte Analysemethode zur Refinanzierung von Struktureinschränkungen mit übereinstimmenden Informationen. Wir demonstrieren den Wert dieser Informationsquellen in umfangreichen ab initio Strukturvorhersageexperimenten mit einem maßgeschneiderten Konformationsraumsuchalgorithmus und einer neuartigen Strukturvorhersagepipeline. Diese Pipeline erreichte beim 11. gemeinschaftsweiten Experiment zur kritischen Bewertung der Proteinstrukturvorhersage (CASP11) den neuesten Stand der Kontakt- und Ab-Initio-Strukturvorhersageleistung.
Mit unserer CLMS-basierten Hybridmethode rekonstruieren wir die Domänenstrukturen von menschlichem Serumalbumin in Lösung und in seiner nativen Umgebung, dem menschlichen Blutserum. Dies ist ein bahnbrechender erster Schritt auf dem Weg zu einer massenspektrometriebasierten ab initio Strukturbestimmungsmethode, die in der Lage ist, die Struktur von Proteinen dort zu untersuchen, wo sie wirklich wichtig ist: In ihrer natürlichen Umgebung, die ihr eigentlicher Wirkungsort ist.
Dezember 2015