Decoy-Based Template Retrieval for Comparative Modeling
Zusammenfassung
Motivation
Weder experimentelle noch rechnergestützte Methoden sind derzeit in der Lage, die Lücke zwischen bekannten Proteinsequenzen und bekannten Proteinstrukturen zu schließen. Während experimentelle Methoden zu arbeits- und zeitaufwändig sind, scheinen computergestützte Methoden vielversprechend zu sein, haben aber auch ihre eigenen Probleme.
Die vergleichende Modellierung kann in der Regel gute Modelle für eine ungelöste Struktur liefern, da es nur eine begrenzte Anzahl von Faltungen gibt und die meisten von ihnen wahrscheinlich bereits bekannt sind. Die besten Ergebnisse werden erzielt, wenn die Sequenzähnlichkeit zur Identifizierung von Vorlagen genutzt wird. Ist dies nicht möglich, können strukturelle Informationen durch Faltenerkennungsmethoden genutzt werden, wenn auch mit einer geringeren Erfolgsquote. Wenn keine Vorlage gefunden werden kann, kann die Ab-Initio-Modellierung verwendet werden. Dabei wird in der approximierten Energiefunktion eines Proteins nach dem globalen Minimum gesucht, von dem angenommen wird, dass es dem nativen Zustand ähnelt. Dieser Ansatz leidet vor allem unter der Weite des Suchraums, der mit jedem Rest exponentiell wächst.
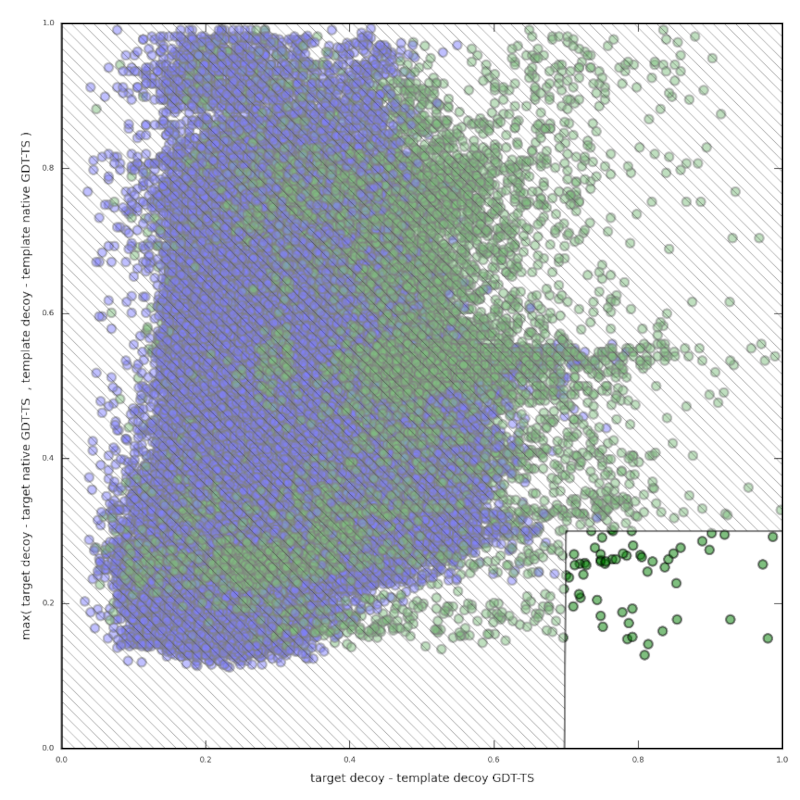
Ziel war es, einen Algorithmus für die Suche nach Vorlagen zu entwickeln, der unabhängig von der Sequenzähnlichkeit ist. Um dies zu erreichen, kombinieren wir ab initio mit vergleichender Modellierung, indem wir nach Ähnlichkeiten in ab initio-Ködern suchen, um gute Vorlagen zu identifizieren. Wir gehen davon aus, dass Decoys eines Targets aus einem von zwei Gründen Ähnlichkeiten mit Decoys von strukturell verwandten Proteinen aufweisen werden. Zum einen können sie einfach deshalb ähnlich sein, weil die Decoys ihren jeweiligen nativen Strukturen ähnlich sind. Diese Ähnlichkeiten können sich bei den Decoys von zwei verschiedenen verwandten Proteinen überschneiden und so die Unterscheidung von Decoys nicht verwandter Proteine ermöglichen. Eine weitere mögliche Ursache ist, dass die Energielandschaften von Proteinen mit verwandten Faltungen ähnliche Minima aufweisen. Dieses Konzept wird durch experimentelle Daten für einen kleinen Bereich der Energielandschaft unterstützt. Es hat sich gezeigt, dass die Übergangszustandsensembles einiger strukturell ähnlicher Proteine auch ähnliche Strukturen aufweisen. Trotz der Beschränkung der experimentellen Daten auf diese spezifische Region könnte dieses Konzept eine größere Allgemeinheit besitzen.
Beschreibung der Arbeit
Die entwickelte Methode generiert ab initio Decoys für alle Targets und einen Satz vorgefilterter Vorlagen und vergleicht sie anhand verschiedener Metriken. Um strukturelle Ähnlichkeiten zwischen den Decoys zu erkennen, bewerten wir die Ähnlichkeit der vorhergesagten strukturellen Motivklassen, der vorhergesagten Kontakte und der Strukturen zwischen den untersuchten Decoys. Anhand der ermittelten strukturellen Ähnlichkeiten erstellten wir eine Rangliste der potenziellen Vorlagen, die wir dann mit einem modernen Tool zur Vorlagenerkennung verglichen.
Ergbeniss
Wir haben die Methode an einer Reihe von 14 Zielen getestet. Jedes Ziel hat 8 - 44 Vorlagen, die von HHsearch abgerufen wurden. Betrachtet man die besten 5% der Vorlagen, die nach den besten Varianten unserer Methode geordnet sind, so wurde die beste Vorlage in 41% der Fälle gefunden.
Die besten 5% der Rangliste von HHsearch enthielten nur in 21% der Fälle die bestmögliche Vorlage. Wir haben herausgefunden, dass die vorgestellte Methode vor allem aufgrund der Ähnlichkeit der Decoys mit den nativen Strukturen erfolgreich ist. Dennoch konnten wir auch Beweise für ähnliche Energielandschaften erbringen, obwohl dieser Gedanke keinen einen großen Einfluss auf die Ergebnisse hatte.